
Loss of Mitochondrial Transcription Factor A Activity Results in Renal Cystic Disease
Ken Ishii, PhD1, Hanako Kobayashi, PhD1, Andraia Li, BS1, Ching Man Carmen Tong, DO2, Olena Davidoff, MS1, Kensei Taguchi, MD/PhD1, Craig Brooks, PhD1, Douglass B. Clayton, MD2, Volker Haase, MD1.
1Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA, 2Division of Pediatric Urology, Monroe Carell Jr. Children's Hospital at Vanderbilt University Medical Center, Nashville, TN, USA.
Introduction
Studies have shown that oxidative stress responses play an important role in early renal cyst formation; specifically, altered mitochondrial protein expression has been demonstrated to be associated with autosomal dominant polycystic kidney disease (ADPKD), one the most common genetic disorders in humans. However, a causal relationship between mitochondrial dysfunction and renal cyst formation has not been clearly established. Here, we demonstrate that Mitochondrial Transcriptional Factor A (TFAM), a key player in mitochondrial DNA replication is required for normal postnatal nephron differentiation and that its genetic ablation leads to the development of cystic phenotype similar to that seen in ADPKD patients.
Methods
We generated a conditional mouse model of renal epithelial TFAM deficiency (Six2-mTmG-TFAM-/-) using the Six2-Cre transgenic mice. We also employed ADPKD mouse models, PKD1-/- and Cyscpk/cpk, to characterize mitochondria with super-resolution Structural Illuminated Microscopy (SIM) and to examine changes in mitochondrial gene expression. Mitochondrial mass and volume were analyzed in these mouse models as well as renal biopsies from PKD human patients. Statistical analyses were performed using the two-tailed Student's t test and ANOVA.
Results
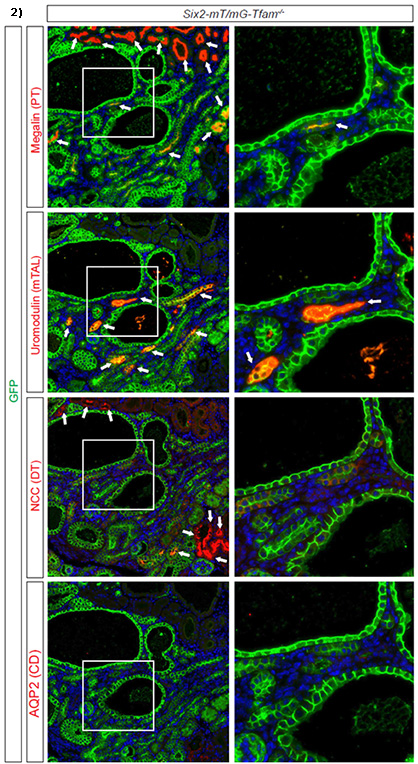
Inactivation of TFAM in epithelial progenitor cells resulted in the development of renal cysts by postnatal day 7 in the Six2-mTmG-TFAM-/- mouse model (denoted # in fig 1), leading to juvenile death. While inactivation of TFAM did not affect nephrogenesis in the embryo, it halted postnatal nephron maturation in TFAM knockout cells as demonstrated by the lack of expression of nephron segment-specific markers, such as Megalin, Uromodulin and Na-Cl cotransporter (fig.2). Importantly, established PKD mouse models confirmed that TFAM is severely downregulated by 90% (p<0.05) along with a 55% decrease in mitochondrial volume on SIM imaging (n=3, p<0.05). Mitochondrial volumes were also significantly decreased in human biopsies between cystic and non-cystic tubules.
Conclusions
Our results collectively support the hypothesis that mitochondrial dysfunction plays an integral role in the formation of renal cysts and maturation of nephrons. Future translational studies will involve investigation of mitochondrial volume and role of TFAM in other renal cystic diseases. 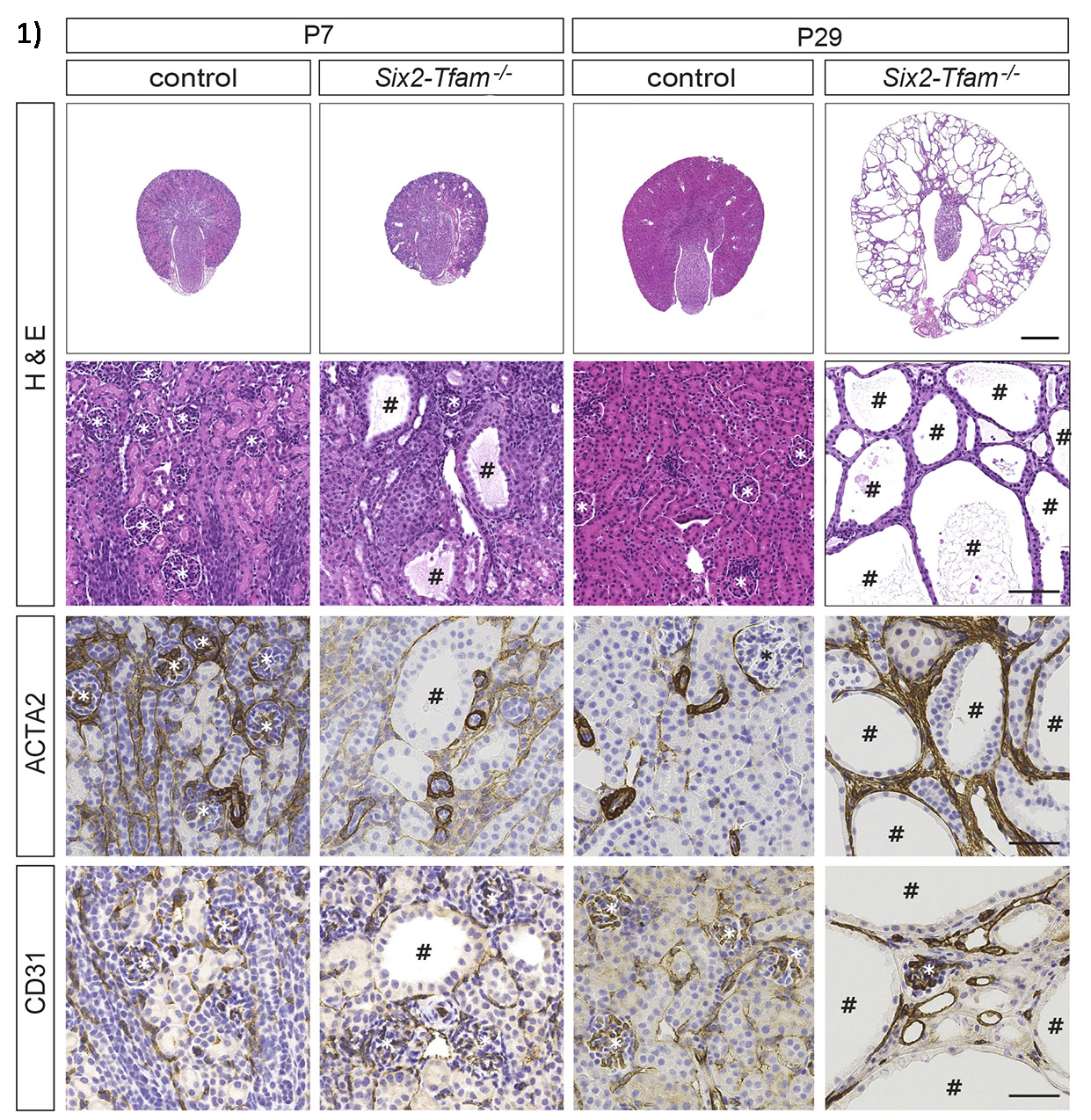
Back to 2019 Abstracts



